Ketone bodies mimic the life span extending properties of caloric restriction
.
Добавление d-βHB к культурам C. elegans увеличивает продолжительность жизни. Мы выдвигаем гипотезу, что увеличение уровней кетоновых тел также продлит срок жизни людей, и что ограничение калорий продлевает продолжительность жизни, по крайней мере частично, за счет увеличения уровней кетоновых тел. Экзогенные кетоновые эфиры представляет собой новый инструмент для подражания эффектам калорийного ограничения, которые могут быть использованы в будущих исследованиях. Возможность питания митохондрий у пожилых людей, которые имеют ограниченную способность к окислению метаболитов глюкозы из-за ингибирования пируватдегидрогеназы, предлагает новые направления исследований для профилактических мер и лечения старения и расстройств, связанных со старением. © 2017 The Authors IUBMB Life published by Wiley Periodicals, Inc. on behalf of International Union of Biochemistry and Molecular Biology, 69(5):305–314, 2017.
Абстракт:
Продление продолжительности жизни калорийным ограничением изучалось по нескольким видам живых организмов от дрожжей и Caenorhabditis elegans до приматов. Для объяснения этих наблюдений не было предложено общепринятой теории. Здесь мы предполагаем, что продление продолжительности жизни, вызванное калорийным ограничением, может быть продублировано метаболическими изменениями, вызванными кетозом.
От нематод до мышей увеличение продолжительности жизни обусловлено снижением передачи сигналов по пути передачи сигналов рецептора инсулина/инсулиноподобного фактора роста (IIS). Снижение IIS уменьшает продукцию фосфатидилинозитола (3,4,5) трифосфата (PIP3), что приводит к снижению активности киназы PI3K и AKT и уменьшению фосфорилирования фактора транскрипционного фактора F (FOXO), что позволяет белкам FOXO оставаться в ядре. В ядре белки FOXO увеличивают транскрипцию генов, кодирующих антиоксидантные ферменты, включая супероксиддисмутазу 2, каталазу, глутатионпероксидазу и сотни других генов.
Эффективным методом борьбы со свободными радикалами является метаболизм кетоновых тел, а кетоз является характерным физиологическим изменением, вызванным ограничением калорийности для живых организмов от плодовых мух до приматов. Диетический кетоновый эфир также снижает циркулирующую глюкозу и инсулин, что приводит к уменьшению IIS. Кетоновое тело, d-β-гидроксибутират (d-βHB), является естественным ингибитором гионановых деацетилаз класса I и IIa, которые подавляют транскрипцию гена FOXO3. Поэтому кетоз приводит к транскрипции ферментов антиоксидантных путей. Кроме того, метаболизм кетоновых тел приводит к более негативному окислительно-восстановительному потенциалу антиоксидантной системы НАДФ (a more negative redox potential of the NADP antioxidant system), которая является системой, уничтожающей свободные радикалы.
Добавление d-βHB к культурам C. elegans увеличивает продолжительность их жизни. Мы выдвигаем гипотезу, что увеличение уровней кетоновых тел также продлит срок жизни людей и что ограничение калорий продлевает продолжительность жизни, по крайней мере частично, за счет увеличения уровней кетоновых тел. Экзогенный кетоновый эфир представляет собой новый инструмент для воспроизведения эффектов калорийного ограничения, которые могут быть использованы в будущих исследованиях. Возможность питания митохондрий у пожилых людей, которые имеют ограниченную способность к окислению метаболитов глюкозы из-за ингибирования пируватдегидрогеназы, предлагает новые направления исследований для профилактических мер и лечения старения и расстройств, связанных со старением. © 2017 The Authors IUBMB Life published by Wiley Periodicals, Inc. on behalf of International Union of Biochemistry and Molecular Biology, 69(5):305–314, 2017.
Кетоновые тела и продление жизни
В 1935 году МакКей и коллеги продемонстрировали своими исследованиями, что ограничение калорий на 30-50% увеличивает средний срок жизни крыс от 500 до 820 дней 1. С тех пор было показано, что калорийное или диетическое ограничение увеличивает продолжительность жизни у самых разных видов: от дрожжей 2 до нематоды 3 до плодовых мух 4 до мышей 5 и приматов. Было показано, что в исследованиях приматов ограничение калорий продлевает продолжительность жизни одной группой 6, но более раннее исследование, использующее несколько иной протокол ограничения калорийности, не оказало влияния на продолжительность жизни 7. Был предложен ряд предложительных механизмов явлений, включая: замедление роста, снижение содержания жира, снижение воспаления, снижение окислительного повреждения, температура тела и сигнализация инсулина, увеличение физической активности и аутофагии 8. Однако для этого широко наблюдаемого явления вообще не было согласованного механистического объяснения, что ограничение калорийности продолжительность жизни по всем видам. Тем не менее, очевидным метаболическим изменением, связанным с ограничением калорийности, является кетоз. Повышенные концентрации кетонового тела происходят во время калорийного ограничения у самых разных видов, начиная от Caenorhabditis elegans 9 и Drosophila 4 до человека, где кетоновые тела вырабатываются в печени из свободных жирных кислот, высвобождаемых из жировой ткани 10.
Кетоновые тела были впервые обнаружены в моче пациентов с диабетом 11, создав у врачей мысль о том, что присутствие кетонов патологично. Однако Кэхилл показал, что кетоновые тела являются нормальным результатом голодания у человека 12, когда они могут использоваться у человека в большинстве внепеченочных тканей, включая мозг 13. Кетоновые тела, d-β-гидроксибутират (d-βHB) и его редокс-партнер ацетоацетат увеличиваются во время голодания 14, физических упражнений 15 или низкоуглеводной диеты 16. Первоначально считалось, что кетоновые тела образуются путем реверсии пути β-окисления жирных кислот. Однако Лехнингер и Гревилль окончательно и изящно показали, что β-гидроксибутират пути β-окисления имеет l-форму, а в процессе кетогенеза — d-форма 17. Это фундаментальное различие в метаболизме d и l формы кетоновых тел имеет глубокие метаболические эффекты.
Метаболизм d-формы приводит к окислению митохондриального co‐enzyme Q 18 и увеличению окислительно-восстановительного промежутка между митохондриальными парами NAD и Q с результирующим увеличением ΔG’ аденозинтрифосфата (АТФ). L-форму β-гидроксибутирата активируют путем превращения АТФ в аденозинмонофосфат (AMP), что является более энергетически дорогостоящим процессом, чем активация сукцинил-СоА. В отличие от метаболизма d-βHB, который продуцирует только NADH, дальнейший метаболизм l-формы метаболизируется системой β-окисления жирных кислот, что приводит к уменьшению одного митохондриального NAD и одного ко-фермента Q с нет увеличения количества окислительно-восстановительного интервала между двумя парами и, следовательно, никакого увеличения ΔG ‘гидролиза АТФ. При катаболизации для синтеза АТФ в митохондриях, d-βHB вырабатывает больше АТФ на молекулу потребляемого кислорода, чем многие другие респираторные субстраты, благодаря этой уникальной природе метаболизма d-βHB 18, 19.
Уровень кетонов в крови также может быть увеличен без повышения содержания свободных жирных кислот путем приема внутрь эфиров кетоновых тел, таких как моноэфир d-βHB-R 1,3 бутандиола 20. Этот кетоновый эфир прошел оценку токсичности 21 и признан как безопасный (GRAS) Управлением по контролю за продуктами и лекарствами (the Food and Drug Administration).
Недавно было показано, что введение d-βHB C. elegans вызвало увеличение продолжительности жизни, в результате чего кетоновые тела предварительно маркировано как «кетоновые тела противостареющего/антивозрастного действия» 9. В том же эксперименте l-β- гидроксибутират не смог продлить срок жизни. Если принято утверждение, что кетоновое тело d-βHB является «антивозрастным» соединением, это может объяснить широко распространенное наблюдение, что ограничение калорийности и результирующий вследствие кетоз приводит к увеличению продолжительности жизни. Многие сигнальные пути, опосредующие продление срока службы, были определены генетиками в основном благодаря работе с недолговечной нематодой C. elegans.
Генетические Механизмы Увеличения продолжительности Жизни
.
Имеются заметные наследуемые различия в средней продолжительности жизни между видами: от менее чем 3 недели у C. elegans, от 2 до 3 лет для мыши или крысы, от 10 до 15 лет для собаки, около 70 лет для людей и до более 400 лет для двустворчатого моллюска Artica islandica. Это наблюдение дает понять, что продолжительность жизни имеет наследуемый компонент.
Сообщалось о первом генетически обусловленном увеличении продолжительности жизни у C. elegans при мутации в age‐1, кодирующей каталитическую субъединицу 3-OH-киназы фосфатидилинозитола рецептора рецептора инсулина/инсулиноподобного фактора роста (IGF) (IIS) 22 23. Лаборатория Kenyon проницательно воспользовалась экспериментальным преимуществом короткой продолжительности жизни C. elegans для идентификации мутаций в гене адренорецептора аномального генерации dauer-2 (daf-2) инсулина/IGF-1 пути IIS, который привел к двукратному увеличение продолжительности жизни C. elegans 24. Было установлено, что это продление продолжительности жизни было главным образом вызвано уменьшением фосфорилирования и ядерной транслокации регулятора транскрипции DAF-16 / FOXO, что привело к экспрессии более 200 генов, в том числе тех, которые участвуют в метаболизме, протеостазе и антиоксидантной защите 25, 26 (фиг.1). Активация других факторов, таких как регулятор транскрипции SKN-1/Nrf2, также способствует повышению долговечности уменьшенного IIS при определенных экспериментальных условиях 27. Увеличение продолжительности жизни мутанта daf-2 может быть дополнительно увеличено с помощью диетического ограничения, указывающего, по меньшей мере, частично различные механизмы действия 28.
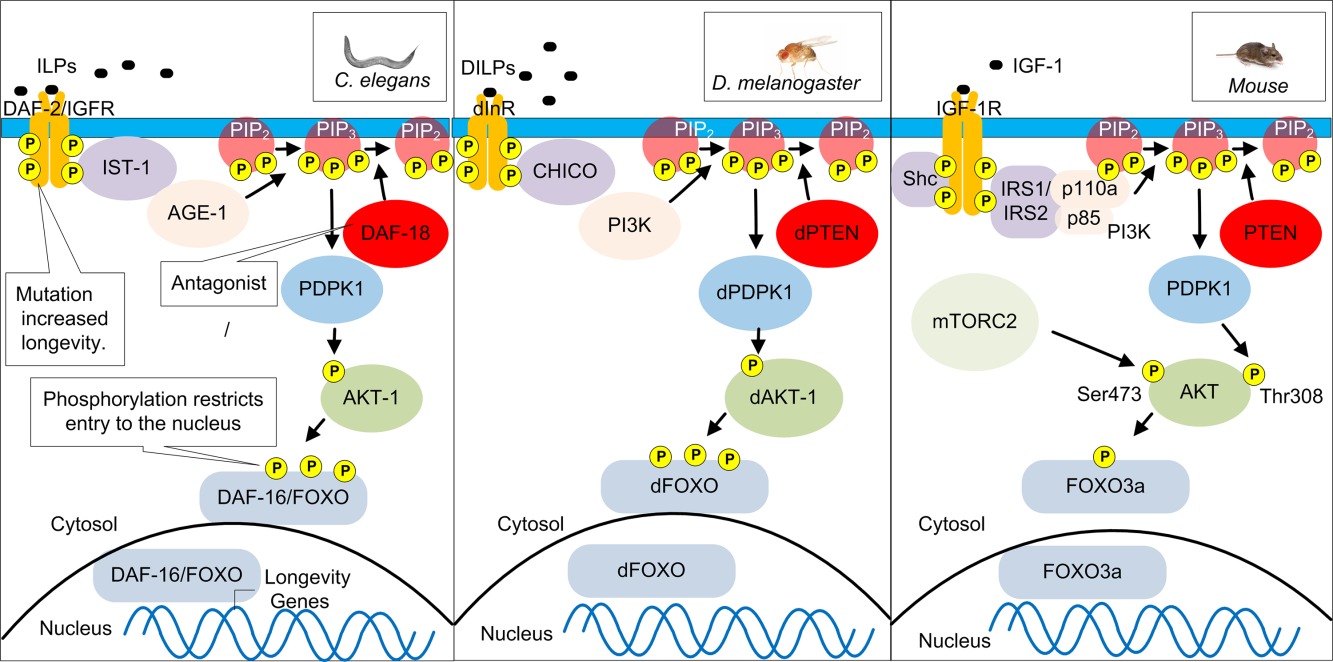
Аббревиатуры: AGE-1 представляет собой фосфоинозитид-3-киназу (PI3K), PIP3 обозначает фосфатидилинозитол (3,4,5) -трифосфат, PIP2 является аббревиатурой для фосфатидилинозита 4, 5-бисфосфата, PTEN представляет собой фосфатазу и гомолог TENsin, PDPK1 представляет собой фосфоинозитид-зависимая киназа-1, AKT названа в отношении штамма мышей Ak, а t обозначает тимому. Дауэр по-немецким означает выносливость или продолжительность, что указывает на приостановленную стадию развития. DAF — это семейство белков, которые были исследованы в качестве дауэр-факторов, которые регулируют вход в состояние дауэра, CHICO означает маленького мальчика по-испански, отсылка к малому размеру тела мутантных мух. mTORC2 представляет собой комплекс, содержащий механическую мишень рапамицина (mTOR). Он состоит из mTOR, Rictor, mLST8, Protor1 / 2, Sin1 и Deptor. FOXO указывает коэффициент транскрипции брандмауэра. ILP является аббревиатурой для инсулиноподобных пептидов. IGF-1 означает инсулиноподобный фактор роста-1.
.
Сначала было непонятно, почему мутация или уменьшенная экспрессия гена рецептора daf-2 инсулина / IGF-1 у C. elegans была связана с продлением срока жизни. В конце концов, IGF вызывает пролиферацию в клетках, которые не постмитотичны и гипертрофию в миоцитах. Ирония в том, что учитывая привлекательность сладкого вкуса, потребление глюкозы с результирующей стимуляцией рецептора инсулина должно быть сигналом для сокращения продолжительности жизни. Пониженная сигнализация через этот путь, вероятно, превратилась в механизм замедления размножения, пока пища не была более обильной, а задержка в воспроизводстве привела к увеличению выживаемости. Более понятным является вывод о том, что увеличение активности белка DAF-16/FOXO, контролирующего экспрессию нескольких антиоксидантных ферментов и белков теплового шока, должно играть ключевую роль в продлении жизненного цикла. Однако, поскольку у животных развились более длительные жизненные периоды, у белков FOXO образовались дополнительные более сложные роли в регулировании клеточной функции и старения, включая стимулирующий апоптоз 29, который, вероятно, помогает предотвратить опухолегенность 30. Белки FOXO модифицируются после трансляционно путем ацетилирования и фосфорилирования, которые регулируются по многим факторам, включая метаболизм, воспаление и окислительный стресс 29.
Сложные роли и механизмы сигнализации, которые, как считается, контролируют экспрессию и активность различных белков FOXO, недавно были пересмотрены 31. Аллель FOXO3а, одного из четырех генов FOXO человека, ассоциируется с экстремальной долговечностью у людей 32. Долголетие можно регулировать через другие специфические сигнальные пути и регуляторы транскрипции, такие как механистическая мишень рапамицина (mTOR), 5′-AMP-активированная протеинкиназа (AMPK), сиртуин 1 (SIRT1), сиртуин 3 (SIRT3), ядерный фактор (2-подобный 2-й эритроид 2) (Nrf2) и активирующий пролифератор активированный пероксисом рецептор 1-α (PGC-1α) 33, хотя ортогены нематод PGC-1α и SIRT3, по-видимому, отсутствуют.
Старение, Окислительный Стресс, и Антиоксидантная Система, Связанная с NADPH
В 1950-х годах Харман предположил, что токсичность реакционноспособных видов кислорода/свободных радикалов (ROS) является центральной для механизма старения 34, поскольку это связано с радиационной токсичностью 35. С тех пор накоплено доказательство того, что механизм, ограничивающий продолжительность жизни, является результатом повреждения ROS 36, 37. Позднее полученные данные 37 значительно поддерживали теорию старения, согласно которой митохондрии повреждались свободными радикалами. Первое из них — сильная обратная корреляция между образованием митохондриальных ROS и долгожительством, а вторая — сильная обратная корреляция между степенью ненасыщенности жирных кислот в тканевых мембранах и долговечностью среди родственных видов. Никакие другие измеренные параметры не соотносились с продолжительностью жизни, которые, вероятно, эволюционировали, чтобы минимизировать ROS-опосредованный ущерб.
.
Хотя ROS и реактивные виды азота (RNS) необходимы для определенных сигнальных путей, их нерегулируемое производство разрушительно и приводит к патологии. Однако за последние 10 лет доказательства, полученные на C. elegans и других модельных животных, продемонстрировали, что для некоторых механизмов продления жизненного цикла требуется опосредованная ROS сигнализация.
.
Токсичность ROS / RNS улучшается системой NADPH (рис. 2), окислительно-восстановительный потенциал которого более отрицателен в результате метаболизма кетоновых тел 19, 38, 39. Редокс-потенциал свободного цитозольного [NADP +] / [NADPH] составляет около -0,42 В, примерно такой же окислительно-восстановительный потенциал, что и у водорода, и является самым негативным окислительно-восстановительным потенциалом в организме 38. Другие восстановители, такие как пара аскорбиновой кислоты, ферментативно связаны с [NADP +] / [ NADPH]. 19. В периферических тканях в сытом состоянии NADPH в основном получают из метаболизма глюкозы по пути гексозомонофосфата 40. Во время голодания, когда глюкоза ограничена, NADPH получают из метаболизма кетоновых тел в цикле Кребса 18, 39 главным образом посредством действия NADP-зависимой изоцитратдегидрогеназы. Во время ограничения калорий митохондриальный SIRT3 деацетилирует и активирует NADP-зависимую изоцитратдегидрогеназу IDH2, что приводит к увеличению продуцирования NADPH и увеличению отношения восстановленного к окислению глутатиона в митохондриях 41. Как FOXO1, так и FOXO3a индуцируют экспрессию IDH1 42, цитоплазматическую форму NADP-зависимая изоцитратдегидрогеназа. Цитрат или изоцитрат, образованный из катаболизма кетонового тела в митохондриях, могут быть экспортированы носителем цитрат-изоцитрат в цитоплазму для получения NADPH по IDH1 (рис.2).
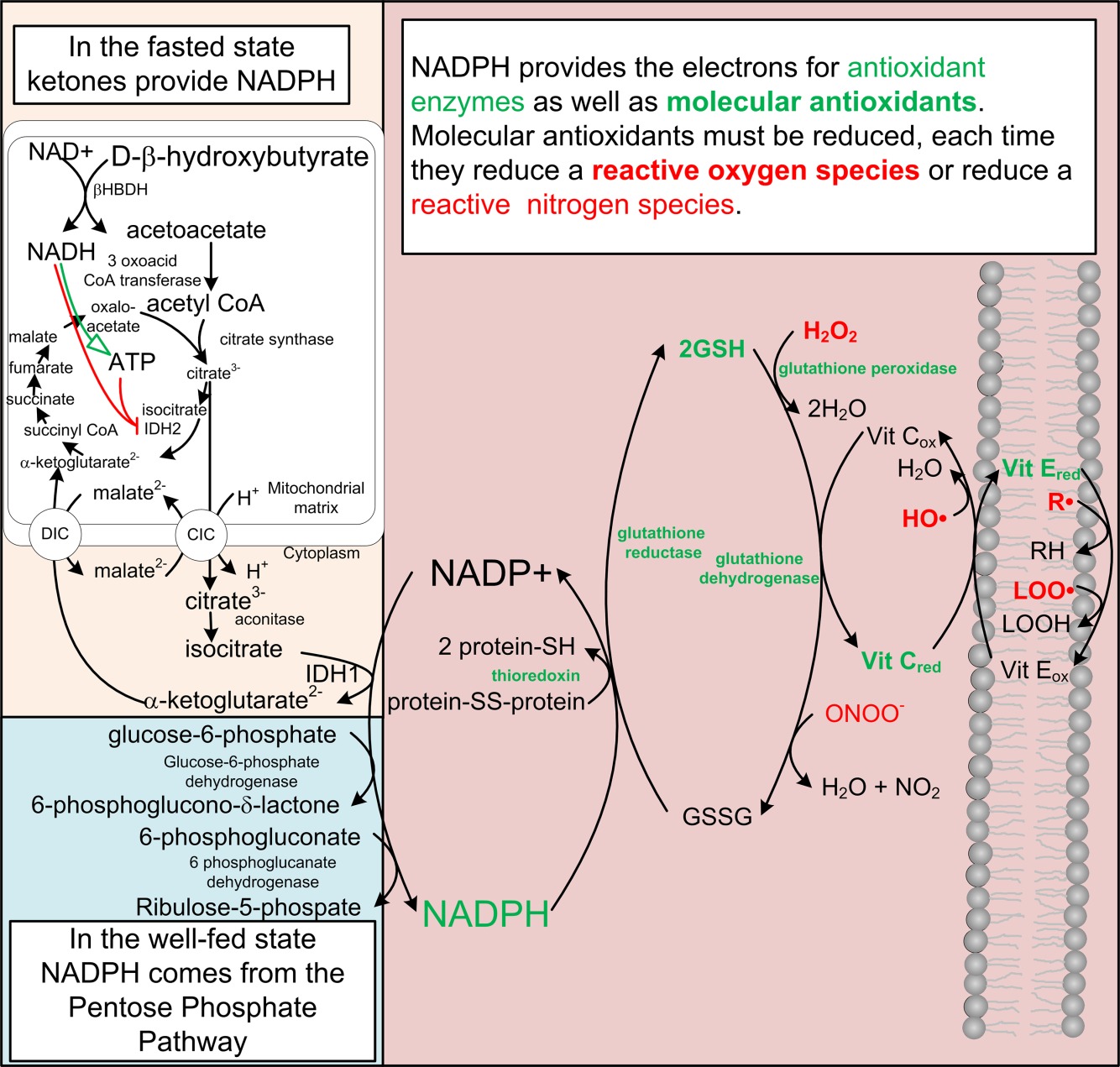
Система NADPH антиоксидантных ферментов и NADPH-зависимые молекулярные антиоксиданты. Показаны два основных пути, обеспечивающие достаточное количество доноров электронов для восстановления окисленных видов в цитозоле, органеллах и мембранах. Это достигается, частично, посредством NADPH-зависимого восстановления глутатиона (GSH), витамина C (Vit C) и витамина E (Vit E). Редокс-потенциал этих вторичных систем определяется окислительно-восстановительным потенциалом системы свободного цитозоля [NADP+]/ [NADPH], с которой они связаны ферментативно.
Addition of reducing agents such as ascorbic acid did not uniformly increase the life span of model organisms, perhaps because these treatments had both pro and antioxidant effects or that reducing agents block ROS signals required for life span extension. Extravagant claims for the beneficial effects of high doses of ascorbic acid made by Linus Pauling were largely unsubstantiated. It was through the work of Krebs and Veech that the control of redox states in the cell and the dominant role of the free [NADP+]/[NADPH] was appreciated 38. The detoxification of free radicals is dependent upon the multiple redox couples which are linked to and whose redox potential is set by the most negative NADP system 19. In the absence of malnutrition, there is little or no reason to take antioxidant supplements because the ability of these compounds to function as antioxidants is largely determined by the [NADP+]/[NADPH]. As aluded to above, this ratio is regulated by the flux of substrates through enzymes that generate or consume NADP+ and NADPH, the reduction of which is brought about by the metabolism of ketone bodies. Therefore, consuming increased amounts of antioxidants has little effect on the [NADP+]/[NADPH].
Добавление восстановителей, таких как аскорбиновая кислота, не равномерно увеличивало продолжительность жизни модельных организмов, возможно, потому, что такое воздействие имело как про-, так и антиоксидантный эффект, или что восстановители блокируют сигналы ROS, необходимые для продления срока жизни. Экстравагантные утверждения о положительном влиянии высоких доз аскорбиновой кислоты, сделанные Линусом Полингом, были в значительной степени необоснованными. Именно благодаря работе Кребса и Вика контроль над окислительно-восстановительными состояниями в клетке и доминирующая роль свободного [NADP+]/[НАДФ] был оценен 38. Детоксикация свободных радикалов зависит от множественных окислительно-восстановительных пар, которые являются связанными с редокс-потенциалом, и его окислительно-восстановительный потенциал установлен самой негативной системой НАДФ 19. В отсутствие недоедания нет никаких оснований принимать антиоксидантные добавки, поскольку способность этих соединений функционировать как антиоксиданты в значительной степени определяется [NADP+], / [НАДФН]. Как упоминалось выше, это соотношение регулируется потоком субстратов через ферменты, которые генерируют или потребляют NADP+ и NADPH, уменьшение которых обусловлено метаболизмом кетоновых тел. Поэтому потребление повышенного количества антиоксидантов мало влияет на [NADP+] / [NADPH].
Data in apparent conflict with the free radical theory of aging has led to its increased scrutiny. In C. elegans, one study showed that increasing free radicals by knocking out the superoxide dismutase (SOD) genes one at a time did not shorten life. Knockout of sod‐4, an extracellular protein, even had the counter‐intuitive effect of extending life span 43. Another group showed that mice engineered to overexpress SOD and catalase did not live longer than normal 44. However, overexpressing mitochondrial‐targeted catalase in mice did lead to life span extension 45. SOD and catalase are both dismutases that are not linked to the NADPH system of clearing ROS/RNS that we hypothesize to be the most important driving force for life span extension.
Данные, явно противоречащие теории старения из-за воздействия свободных радикалов, привели к ее тщательному пересмотру. В случае C. elegans одно исследование показало, что увеличение количества свободных радикалов путем выбивания генов супероксиддисмутазы (SOD) по одному не сокращало жизнь. Knockout of sod‐4 внеклеточного белка даже имел противоинтуитивный эффект продления жизненного цикла 43. Другая группа показала, что мыши, созданные для сверхэкспрессии SOD, и каталаза не живут дольше, чем обычно 44. Однако сверхэкспрессирующая митохондриальная каталаза у мышей действительно приводили к увеличению продолжительности жизни 45. SOD и каталаза — это оба дисмутазы, которые не связаны с системой очистки ROS / RNS NADPH, которые, по нашим предположениям, являются наиболее важной движущей силой продления срока жизни. Имеются существенные данные, подтверждающие способность уменьшать [NADP+] / [НАДФ] для увеличения продолжительности жизни.
There is substantial data supporting the ability of decreased [NADP+]/[NADPH] to extend life span. Glucose‐6‐phosphate dehydrogenase overexpression increased median life span in Drosophila 46 and female mice 47. Longer‐lived strains of Drosophila were shown to possess higher glucose‐6‐phosphate dehydrogenase activity than shorter lived strains 48. By increasing flux through NADP‐dependent forms of the enzyme, knocking out or knocking down NAD‐dependent isocitrate dehydrogenase increased life span in yeast 49 and C. elegans 50. Finally, overexpression of NADP‐dependent malic enzyme extended life span in Drosophila 51. Studies should now be performed to more directly test the hypothesis that increased NADPH levels extend lifespan through bolstering the NADPH antioxidant system.
Существует большое колличество данных, что избыточная экспрессия глюкозо-6-фосфатдегидрогеназы увеличивает средний срок жизни у Drosophila 46 и самок мышей 47. Показано, что более длительные штаммы Drosophila обладают более высокой активностью глюкозо-6-фосфатдегидрогеназы, чем более короткоживущие штаммы 48. Увеличивая поток через NADP-зависимую формы фермента, выключение или включение NAD-зависимой изоцитратдегидрогеназы увеличивает продолжительность жизни у дрожжей 49 и C. elegans 50. Наконец, чрезмерная экспрессия NADP-зависимого развития яблочного фермента увеличивает продолжительность жизни у Drosophila 51. Теперь исследования должны быть выполнены более непосредственно проверяют гипотезу о том, что увеличение уровней NADPH продлевает срок службы путем укрепления антиоксидантной системы NADPH.
Метаболизм Кетоновых Тел Позволяет Обойти Снижение Активности Пируватдегидрогеназы в Стареющих Тканях Тела
Decreased Pyruvate Dehydrogenase Activity in Aged Tissues is Bypassed by Ketone Body Metabolism
Aging has been shown to lead to decreased mitochondrial pyruvate dehydrogenase (PDH) complex activity. In heart, this decreased activity was not due to lower PDH complex levels, but due to increased phosphorylation that inhibits complex activity 52. PDH phosphatases, which are able to increase PDH activity, have been shown to be stimulated by insulin 53, and in skeletal muscle, this stimulation was shown to decline with age, but be restored by exercise 54. Decreased PDH activity has been found in specific regions of aged brain such as in the striatum and brain stem as a result of increased PDH kinase activity 55. This finding could result from increased ROS production from mitochondria during aging that increases hypoxia‐inducible factor 1‐alpha (HIF‐1α) activity 56 and the expression of the pyruvate dehydrogenase kinase isoform 4 (PDK4) 57. Consistent with it playing a role in the aging process, the PDH complex has also been shown to regulate cellular senescence 58. However, too much PDH activity may have pro‐aging effects through a hyper‐stimulation of mitochondrial metabolism resulting in increased ROS production. Therefore, FOXO proteins have evolved to induce expression of PDH kinases such as PDK4 to negatively regulate PDH activity and ROS production 59. During fasting and caloric restriction, the FOXO‐mediated expression of PDK enzymes may also serve an important role in the shunting of pyruvate and/or lactate to cell types or tissues with the highest energy needs, such as neurons that cannot oxidize fatty acids.
Показано, что старение приводит к снижению активности комплекса митохондриальной пируватдегидрогеназы (PDH). В сердце эта пониженная активность не была вызвана более низкими уровнями PDH-комплекса, а из-за повышенного фосфорилирования, которое ингибирует комплексную активность 52. Показано, что фосфатазы PDH, способные повышать активность PDH, стимулируются инсулином 53, а в скелетных мышцы, эта стимуляция, как было показано, снижается с возрастом, но восстанавливается посредством упражнений 54. Снижение активности PDH было обнаружено в определенных областях престарелого мозга, таких как стриатум и мозговой шток, в результате повышенной активности киназы PDH 55. Это открытие может приводить к увеличению продукции ROS из митохондрий во время старения, что увеличивает активность индуцируемого гипоксией фактора 1-альфа (HIF-1α) 56 и экспрессию изоформы 4 пируватдегидрогеназы киназы (PDK4) 57. В соответствии с этим он играет роль в старении процесс, PDH-комплекс также, как было показано, регулирует клеточное старение 58. Однако слишком большая активность PDH может оказывать про-стареющее действие посредством гиперстимуляции митохондриального метаболизма ism, что приводит к увеличению производства ROS. Поэтому белки FOXO эволюционировали, чтобы индуцировать экспрессию PDH-киназ, таких как PDK4, чтобы отрицательно регулировать активность PDH и продукцию ROS 59. Во время голодания и ограничения калорий, FOXO-опосредованная экспрессия ферментов PDK может также играть важную роль в шунтировании пирувата и / или лактат к типам клеток или тканям с наивысшими потребностями в энергии, таким как нейроны, которые не могут окислять жирные кислоты.
Studies in C. elegans also support a role for PDH activity in the regulation of longevity. Inhibiting PDH kinase activity with dichloroacetate extended life span 60, while overexpression of the PDH phosphatase, PDP‐1, also increased longevity through increased DAF‐16/FOXO nuclear translocation 61. It is important to determine whether PDH activity regulates the activity of FOXO proteins in humans. This could potentially occur through nuclear localized PDH providing acetyl‐CoA for histone acetylation 62 of FOXO promoters. If PDH activity stimulates FOXO expression, declining PDH activity during aging may lead to a downstream loss of FOXO‐mediated transcriptional events and increased oxidative stress. As metabolism of ketone bodies bypasses PDH activity as shown in Fig. 3, ketone or ketone ester supplementation may be able to mitigate metabolic and transcriptional alterations resulting from decreased PDH activity to promote longevity. Calorie restriction was shown to stimulate skeletal muscle mitochondrial pyruvate metabolism by increasing expression of the mitochondrial pyruvate carrier and decreasing expression of the lactate dehydrogenase A gene 63. The longevity effects of caloric restriction in mammals require the FOXO3a gene 64.
Исследования C. elegans также поддерживают роль активности PDH в регуляции долголетия. Ингибирование активности киназы PDH с увеличением продолжительности жизни дихлорацетата 60, в то время как избыточная экспрессия PDH-фосфатазы PDP-1 также увеличивала долговечность за счет увеличения транслокации DAF-16 / FOXO. 61. Важно определить, регулирует ли активность PDH активность белков FOXO в людях. Это может потенциально происходить через ядерный локализованный PDH, обеспечивающий ацетил-CoA для ацетилирования гистонов 62 промоторов FOXO. Если активность PDH стимулирует экспрессию FOXO, снижение активности PDH во время старения может привести к снижению активности транскрипционных событий, опосредованных FOXO, и увеличению окислительного стресса. Поскольку метаболизм кетоновых тел обходит активность PDH, как показано на рис.3, добавление кетонового или кетонового эфира может способствовать уменьшению метаболических и транскрипционных изменений, вызванных сниженной активностью PDH для повышения долговечности. Было показано, что ограничение калорий стимулирует метаболизм митохондриального пирувата скелетных мышц за счет увеличения экспрессии митохондриального пируватного носителя и уменьшения экспрессии гена лактатдегидрогеназы А 63. Эффект долговечности калорийного ограничения у млекопитающих требует ген FOXO3a 64.

Рисунок 3
Ketone body metabolism bypasses decreased PDH activity in aged tissues.
Кетоновый метаболизм тела позволяет компенсировать (?) снижение активности PDH в престарелых тканях.
Telomere Shortening is Linked to Cellular Redox Status and Metabolism
Укорачивание теломер связано со статусом и метаболизмом клеточной редокс системы
The work of Hayflick and Moorhead 65 pointed out that shortening of the telomeres set a limit to the number of divisions cells in culture could undergo before senescence occurs. Expression of the telomerase enzyme in certain germ and progenitor cells provides a solution to replicate the ends of linear chromosomes, so that the chromosomes do not become shorter with each new round of DNA replication. Telomeres are lengthened by starvation 66 and shortened by ROS damage 59. These observations are consistent with aging being a function of reactive oxygen and its reversal a function of the increasing redox potential of the NADPH system brought about by caloric restriction. The FOXO protein FOXO1 was shown to be essential for the calorie restriction‐mediated increase in telomerase subunit expression 67. As cells approach their Hayflick limit, the expression of the FOXO genes FOXO1 and FOXO4 have also been shown to decline 68, which would lead to decreased SOD2 and catalase expression. Senescent cells and tissues not only show decreased function but also acquire a senescence‐associated secretory phenotype (SASP), a pro‐inflammatory, pro‐aging state. Mitochondrial dysfunction that increases ROS/RNS production also induces a cellular senescence program with a modified SASP 67.
В работе Hayflick и Moorhead 65 указывалось, что укорочение теломер, ограничивающих количество клеток деления в культуре, может пройти до старения. Экспрессия фермента теломеразы в некоторых клетках зародыша и предшественника обеспечивает решение для репликации концов линейных хромосом, так что хромосомы не становятся короче с каждым новым циклом репликации ДНК. Теломеры удлиняются голодом 66 и укорачиваются повреждением ROS 59. Эти наблюдения согласуются со старением, являющимся функцией реактивного кислорода и его изменением функции возрастающего окислительно-восстановительного потенциала системы НАДФН, вызванной калорийным ограничением. Было показано, что белок FOXO FOXO1 необходим для опосредуемого калорией увеличения экспрессии 67 субъединицы теломеразы. Поскольку клетки приближаются к пределу Hayflick, также было показано, что экспрессия FOXO-генов FOXO1 и FOXO4 снижается на 68, что приведет к снижение SOD2 и экспрессию каталазы. Сенсибилизированные клетки и ткани не только проявляют пониженную функцию, но также приобретают связанный с старением секреторный фенотип (SASP), провоспалительное, про-стареющее состояние. Митохондриальная дисфункция, которая увеличивает продукцию ROS / RNS, также вызывает программу клеточного старения с модифицированным SASP 67.
Other Potential Anti‐Aging Mechanisms of Ketone Bodies
Decreased insulin signaling activates mammalian FOXO proteins such as FOXO1 and FOXO3, which stimulates expression of many genes involved in autophagy 69. In addition, decreased insulin signaling or nutrient deprivation inactivates mTOR kinase to stimulate autophagy, which is required for dietary restriction‐mediated life span extension in C. elegans 70. Consistent with these effects, d‐βHB treatment has been shown to stimulate autophagy in cultured cortical neurons 71 and increased rates of autophagy are likely one of the several molecular mechanisms that contribute to the life span extending effects exerted by ketone bodies. One mechanism through which d‐βHB may decrease IIS to activate FOXO proteins and autophagy is through a direct inhibition of AKT 72. This inhibition may also be responsible for the fasting‐induced insulin resistance observed in muscle, heart, and other peripheral tissues that preserves glucose use for the brain 73, 74.
Другие Возможные Механизмы Кетоновых Тел, Препятствующие Старению
Снижение передачи сигналов инсулина активирует белки FOXO млекопитающих, такие как FOXO1 и FOXO3, что стимулирует экспрессию многих генов, участвующих в аутофагии 69. Кроме того, снижение передачи сигналов инсулина или лишение питательных веществ инактивирует киназу mTOR для стимуляции аутофагии, которая требуется для жизни, в C. elegans 70. В соответствии с этими эффектами показано, что лечение d-βHB стимулирует аутофагию в культивируемых кортикальных нейронах 71, а увеличение скорости аутофагии, вероятно, является одним из нескольких молекулярных механизмов, которые вносят вклад в расширение продолжительности жизни, оказываемое кетоновыми телами. Один из механизмов, посредством которых d-βHB может уменьшить IIS для активации белков FOXO и аутофагии, заключается в прямом ингибировании AKT 72. Это ингибирование также может быть ответственным за индуцированную натощак резистентность к инсулину, наблюдаемую в мышцах, сердце и других периферических тканях, которые сохраняют использование глюкозы для мозга 73, 74.
d‐βHB may also exert protective metabolic effects by binding at least two different G protein‐coupled receptors, HCAR2/Gpr109/PUMA‐G (first discovered to be a nicotinic acid receptor) and free fatty acid receptor 3 on the plasma membrane 75. As these genes evolved in chordates and are not present in invertebrates, they could not function in the evolutionarily conserved role of ketone bodies in life span extension. But activation of these receptors likely plays important metabolic signaling roles mediated by d‐βHB. Finally, the gut microbiome plays an important role in providing substrates for ketone body synthesis 76 and could therefore effect the extent of ketone body synthesis during caloric restriction to influence the magnitude of life span extension, but a further discussion of this research topic is beyond the scope of this review.
d-βHB также может оказывать защитное метаболическое действие путем связывания по меньшей мере двух различных рецепторов, связанных с белком G, HCAR2 / Gpr109 / PUMA-G (впервые обнаруженный как рецептор никотиновой кислоты) и рецептора 3 свободной жирной кислоты на плазматической мембране 75. Поскольку эти гены развивались в хордах и не присутствовали у беспозвоночных, они не могли функционировать в эволюционно сохраняющейся роли кетоновых тел в расширении жизненного цикла. Но активация этих рецепторов, вероятно, играет важную роль метаболической передачи сигналов, опосредованную d-βHB. Наконец, микробиома кишечника играет важную роль в обеспечении субстратов для синтеза кетонового тела 76 и, следовательно, может влиять на объем синтеза кетоновых тел во время ограничения калорийности, чтобы влиять на величину продления жизненного цикла, но дальнейшее обсуждение этой темы исследования выходит за рамки сфера обзора.
Feeding Ketone EstersP
Прием Внутрь Кетоновых Эфиров
One effect of feeding rats with the ketone ester, d‐βHB‐R 1,3 butanediol monoester, was a 1.7‐fold decrease in blood glucose and over a twofold decrease in blood insulin 77. The same decreases in glucose and insulin are seen after feeding ketone esters to mice 78. These metabolic changes induced by feeding ketone esters mimic the decreased IIS induced by a longevity‐inducing mutation in daf‐2 in the nematode 23, 26. In addition to mutations in daf‐2 that increase nuclear translocation and activity of DAF‐16/FOXO, there are ways to increase the transcription of FOXO genes metabolically. For example, in mammals, the transcription of FOXO3a can be induced by inhibition of class I and IIa histone deacetylases (HDACs) by d‐βHB 79 or possibly by β‐hydroxybutyrylation 80 (Fig. 4). Inhibition of these HDACs by d‐βHB induces the expression of other antioxidant and detoxification genes such as the metallothionein‐1 (MTL1) that can lead to reduced ROS toxicity. In liver, fasting also increases FOXO1 activity through a mechanism where glucagon stimulates class IIa HDAC translocation to the nucleus to recruit the class I HDAC HDAC3 to deacetylate FOXO1 81 These pathways also affect metabolism, phosphorylation potential, redox states, and the ability to clear ROS. In C. elegans, the life span extension induced by administration of the ketone body d‐βHB required nematode homologs of AMPK, SIRT1, FOXO, and Nrf2. No additional increase in life span was observed for d‐βHB treatment to a long‐lived S6 kinase mutant of the target of rapamycin (TOR) signaling pathway suggesting that TOR inhibition also plays a role in the ketone body‐mediated longevity effects 33.
Один из эффектов кормления крыс кетоновым эфиром-бетандиолом d-βHB-R 1,3, был в 1,7-кратном снижении уровня глюкозы в крови и более чем двукратным снижением уровня инсулина в крови 77. То же самое снижается при глюкозе и инсулине после кормление кетоновых эфиров мышам 78. Эти метаболические изменения, вызванные питанием кетоновых эфиров, имитируют уменьшенный ИИС, индуцированный мутацией, вызывающей долговечность у нематоды daf-2 23, 26. В дополнение к мутациям у daf-2, которые увеличивают ядерную транслокацию и активности DAF-16 / FOXO, есть способы увеличить транскрипцию генов FOXO метаболически. Например, у млекопитающих транскрипция FOXO3a может быть индуцирована ингибированием гистондезацетилаз класса I и IIa (HDACs) с помощью d-βHB 79 или, возможно, посредством β-гидроксибутирилата 80 (рис.4).
Ингибирование этих HDAC d-βHB индуцирует экспрессию других антиоксидантных и детоксикационных генов, таких как металлотионеин-1 (MTL1), которые могут приводить к снижению токсичности ROS. В печени голодание также увеличивает активность FOXO1 через механизм, при котором глюкагон стимулирует транслокацию HDAC класса IIa к ядру, чтобы набрать HDAC3 класса HDAC3 для деацетилирования FOXO1 81. Эти пути также влияют на метаболизм, потенциал фосфорилирования, окислительно-восстановительные состояния и способность очищать ROS , У C. elegans расширение продолжительности жизни, индуцированное введением кетонового тела d-βHB, требовало нематодных гомологов AMPK, SIRT1, FOXO и Nrf2. Дополнительное увеличение продолжительности жизни не наблюдалось при воздействии d-βHB на долгоживущего мутанта S6-киназы мишени пути передачи сигналов рапамицина (TOR), что указывает на то, что торможение TOR также играет роль в эффектах долголетия, вызванных кетоном.
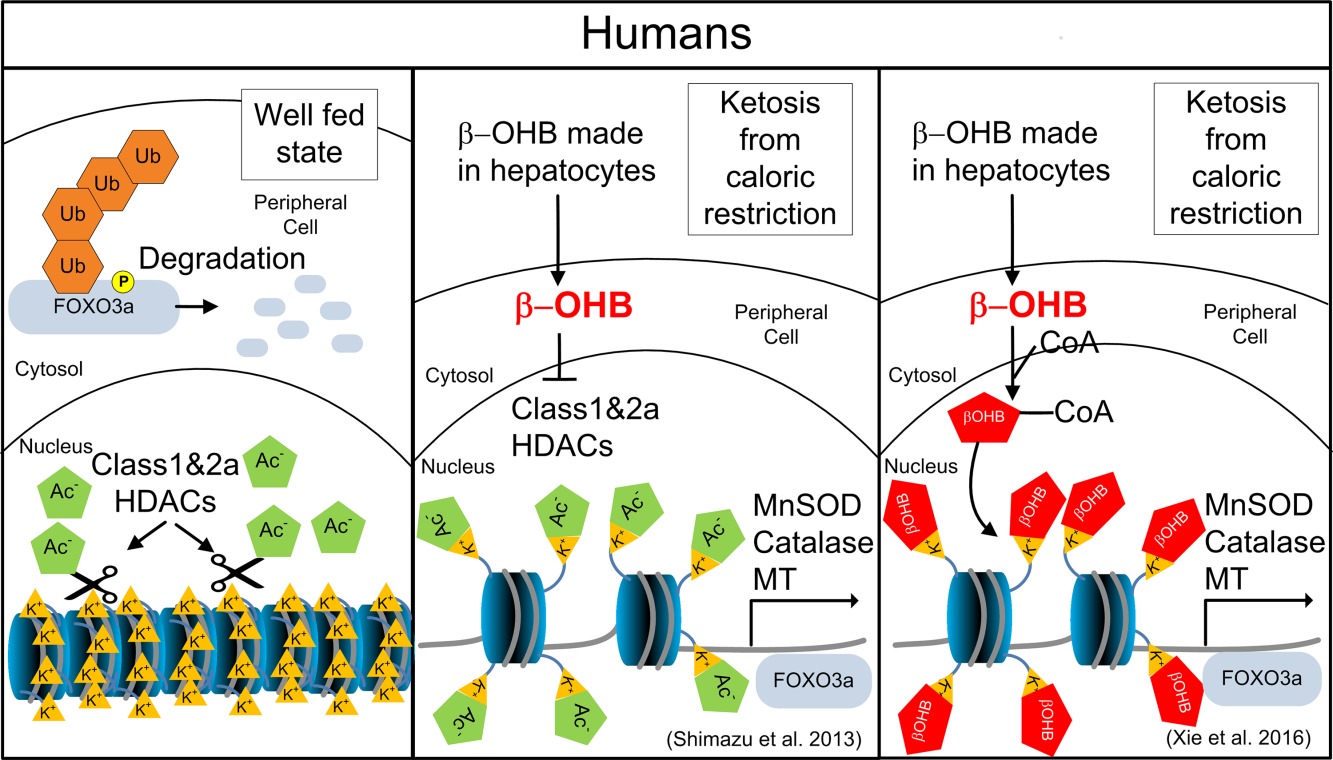
Figure 4
In the well‐fed state, the FOXO3a transcription factor is prevented from entering the nucleus by phosphorylation. FOXO3A is marked for degradation by ubiquitin (Ub). DNA with FOXO3a promoter remains out of reach due to lysine (K+) interacting with negative charges in DNAs phosphodiester backbone keeping histones in the condensed state. In a state of ketosis, HDAC is inhibited by d‐β‐hydroxybutyrate. The acetyl (Ac‐) group neutralizes the charge on lysine opening the histone complex exposing the FOXO3a promoter and upregulating superoxide dismutase (MnSOD), catalase, and metallothionein MT. An alternative mechanism proposed by Xie et al. is shown in the third panel.
В сытом состоянии фактор транскрипции FOXO3a не может попасть в ядро путем фосфорилирования. FOXO3A помечается для деградации убиквитином (Ub). ДНК с промотором FOXO3a остается вне досягаемости из-за взаимодействия лизина (K +) с отрицательными зарядами в фосфодиэфирной цепи ДНК, содержащей гистоны в конденсированном состоянии. В состоянии кетоза HDAC ингибируется d-β-гидроксибутиратом. Ацетил (Ac-) группа нейтрализует заряд на лизин, открывая гистоновый комплекс, подвергая промотор FOXO3a и активирующую супероксиддисмутазу (MnSOD), каталазу и металлотионеин MT. Альтернативный механизм, предложенный Xie et al. отображается на третьей панели.
Несколько отличаясь от генетической манипуляции IIS-пути, подача кетоновых тел приводит к уменьшению отношения свободного цитозольного [NADP +] / [НАДФН] 39, которое обеспечивает термодинамическую силу, необходимую для уменьшения глутатиона и других антиоксидантных пар, которые разрушают свободные от кислорода радикалы 19. Метаболизм кетоновых тел, которые снижают уровень глюкозы в крови и инсулина, снижают активность пути IIS, что в свою очередь приводит к увеличению уровня и активности нефосфорилированных факторов транскрипции FOXO, центральных для расширения продолжительности жизни 26. Кетоз, который является общим следствием ограничения калорийности, может дать объяснение, почему ограничение калорийности приводит к увеличению продолжительности жизни у большинства видов. Здесь мы предполагаем, что продление продолжительности жизни, вызванное калорийным ограничением, может быть продублировано метаболическими изменениями, вызванными кетозом.
Заключение
Старение человека сопровождается ухудшением работы ряда систем. Наиболее заметными являются постепенное повышение уровня сахара в крови и липидов в крови, увеличение сужения кровеносных сосудов, увеличение заболеваемости злокачественными новообразованиями, ухудшение и потеря эластичности кожи, потеря мышечной силы и физиологических физических нагрузок, ухудшение памяти и когнитивных способностей, и у мужчин снижается эректильная функция.
Aging in man is accompanied by deterioration of a number of systems. Most notable are a gradual increase in blood sugar and blood lipids, increased narrowing of blood vessels, an increase in the incidence of malignancies, the deterioration and loss of elasticity in skin, loss of muscular strength and physiological exercise performance, deterioration of memory and cognitive performance, and in males decreases in erectile function.
Было показано, что многие изменения, вызванные старением, такие как заболеваемость злокачественными новообразованиями у мышей 82, увеличение уровня глюкозы в крови и инсулина, вызванное резистентностью к инсулину 39, 78 и мышечной слабостью, снижаются за счет метаболизма кетоновых тел 18, 83 , это нормальный метаболит, полученный из жирных кислот печенью в периоды длительного голодания или ограничения калорийности 12.
Many aging‐induced changes, such as the incidence of malignancies in mice 82, the increases in blood glucose and insulin caused by insulin resistance 39, 78, and the muscular weakness have been shown to be decreased by the metabolism of ketone bodies 18, 83, a normal metabolite produced from fatty acids by liver during periods of prolonged fasting or caloric restriction 12.
Уникальная способность кетоновых тел подавать энергию мозгу в периоды ухудшения метаболизма глюкозы делает кетоз эффективным средством лечения ряда неврологических состояний, которые в настоящее время не имеют эффективных методов лечения. Было также показано, что ухудшение когнитивной функции улучшается за счет метаболизма кетоновых тел 84.
The unique ability of ketone bodies to supply energy to brain during periods of impairment of glucose metabolism make ketosis an effective treatment for a number of neurological conditions which are currently without effective therapies. Impairment of cognitive function has also been shown to be improved by the metabolism of ketone bodies 84.
Кроме того, болезнь Альцгеймера, основной причиной которой является старение 20, может быть улучшена клинически путем введение мягкого кетоза в мышиной модели заболевания 85 и у людей 86. Кетоз также улучшает функционирование при болезне Паркинсона 87, которая, как полагают, в значительной степени вызвана повреждением митохондрий свободныими радикалами 19, 88. Кетоновые тела также полезны для улучшения симптомов бокового амиотрофического склероза 89. Также признано, что кетоз может иметь важные терапевтические применения при широком спектре других заболеваний 90, включая дефицит Glut 1, диабет I типа 91, ожирение 78, 92 и резистентность к инсулину 20, 39, 93 и болезни различной этиологии 90.
Additionally, Alzheimer’s disease, the major cause of which is aging 20 can be improved clinically by the induction of mild ketosis in a mouse model of the disease 85 and in humans 86. Ketosis also improves function in Parkinson’s disease 87 which is thought to be largely caused by mitochondrial free radical damage 19, 88. Ketone bodies are also useful in ameliorating the symptoms of amyotrophic lateral sclerosis 89. It is also recognized that ketosis could have important therapeutic applications in a wide variety of other diseases 90 including Glut 1 deficiency, type I diabetes 91, obesity 78, 92, and insulin resistance 20, 39, 93, and diseases of diverse etiology 90.
В дополнение к улучшению ряда заболеваний, связанных со старением, общее ухудшение клеточных систем, не зависящих от конкретного заболевания, похоже, связано с токсичностью ROS и неспособностью бороться с ним. Напротив, увеличение продолжительности жизни происходит по ряду видов с уменьшением функции пути ИИС и / или активацией факторов транскрипции FOXO, индуцируя экспрессию ферментов, необходимых для детоксикации свободных радикалов (рис.1 и 2). В C. elegans эти результаты были выполнены с использованием РНК-интерференции или мутантных животных. Подобные изменения должны быть достигнуты у высших животных, включая людей, путем введения самого d-βHB или его сложных эфиров.
In addition to ameliorating a number of diseases associated with aging, the general deterioration of cellular systems independent of specific disease seems related to ROS toxicity and the inability to combat it. In contrast increases in life span occur across a number of species with a reduction in function of the IIS pathway and/or an activation of the FOXO transcription factors, inducing expression of the enzymes required for free radical detoxification (Figs. 1 and 2). In C. elegans, these results have been accomplished using RNA interference or mutant animals. Similar changes should be able to be achieved in higher animals, including humans, by the administration of d‐βHB itself or its esters.
Таким образом, снижение передачи сигналов через путь рецептора инсулина / IGF-1 увеличивает продолжительность жизни. Снижение активации рецептора инсулина / IGF-1 приводит к уменьшению PIP3, снижению фосфорилирования и активности фосфоинозитид-зависимой протеинкиназы (PDPK1), снижению фосфорилирования и активности AKT и последующему снижению фосфорилирования FOXO, позволяя им оставаться в ядре и увеличивать транскрипцию ферментов антиоксидантного пути.
У млекопитающих многие из этих изменений могут быть обеспечены метаболизмом кетоновых тел. Метаболизм кетонов снижает уровень глюкозы в крови и уровень инсулина, что снижает активность IIS и его сопутствующие изменения в описанном выше пути. Однако, кроме того, кетоновые тела действуют как естественный ингибитор HDAC класса I, вызывая экспрессию гена FOXO, стимулируя синтез антиоксидантных и метаболических ферментов. Еще одним важным фактором является то, что метаболизм кетоновых тел у млекопитающих увеличивает восстановительную способность системы НАДФ (NADP), обеспечивая термодинамический привод для разрушения свободных радикалов кислорода, которые являются основной причиной процесса старения.
In mammals, many of these changes can be brought about by the metabolism of ketone bodies. The metabolism of ketones lowers the blood glucose and insulin thus decreasing the activity of the IIS and its attendant changes in the pathway described above. However, in addition ketone bodies act as a natural inhibitor of class I HDACs, inducing FOXO gene expression stimulating the synthesis of antioxidant and metabolic enzymes. An added important factor is that the metabolism of ketone bodies in mammals increases the reducing power of the NADP system providing the thermodynamic drive to destroy oxygen free radicals which are a major cause of the aging process.
