A Different Perspective on the Treatment of Type 2 Diabetes
Когда все нормальные жировые отсеки заполнены до отказа, а поступающий жир, содержащий диетический жир и жир, полученный из-за избыточного потребления углеводов, по-прежнему требует места для хранения, возникает побочный эффект. Жир, хранящийся в тех местах, которые никогда не предн азначались для хранения жира, называется эктопическим жиром, т. е. это жир там, где его не должно быть. Различные органы по всему телу, особенно в брюшной полости, поглощают такой избыток жира, а это процесс не без последствий. Во-первых, организм реагирует на этот эктопический жир почти как на инородное тело и мобилизует врожденную иммунную систему для его атаки.
азначались для хранения жира, называется эктопическим жиром, т. е. это жир там, где его не должно быть. Различные органы по всему телу, особенно в брюшной полости, поглощают такой избыток жира, а это процесс не без последствий. Во-первых, организм реагирует на этот эктопический жир почти как на инородное тело и мобилизует врожденную иммунную систему для его атаки.
Макрофаги направляются в эктопический жир и, попав туда, высвобождают воспалительные цитокины и другие аттрактанты для привлечения еще большего количества макрофагов. Объем эктопического жира часто состоит на 50 процентов из макрофагов (3). Во-вторых, эктопический жир может создавать нарушения в различных органах, нарушая их нормальное функционирование.

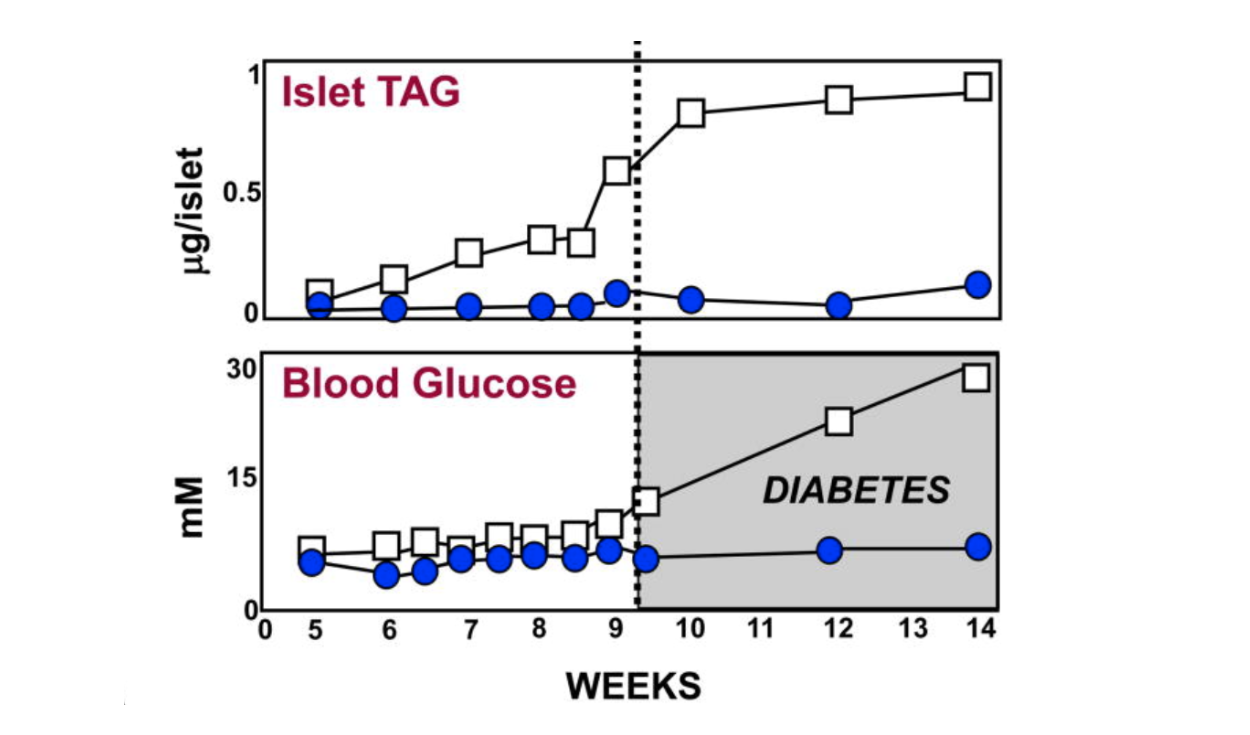
Рисунок 1: Содержание жира в поджелудочной железе у мышей, склонных к диабету (db / db), и худых мышей дикого типа (+ / +)
Но происходит ли это и у людей? На рисунке ниже видно, что это происходит. Ильдико Лингвай и соавторы (Ildiko Lingvay et al.) использовали магнитно-резонансную спектроскопию, чтобы определить содержание жира в поджелудочной железе у людей и сравнить этот показатель со степенью непереносимости глюкозы. Субъектов с нормальной массой тела, устойчивых к глюкозе, сравнивали с субъектами с ожирением, толерантными к глюкозе и ожирением, не переносящими глюкозу. Как видно, при переходе от ожирения к толерантной к глюкозе ситуации, к ожирению с непереносимостью глюкозы наблюдалось значительное увеличение жира в поджелудочной железе (9). Даже субъекты с полностью развитым СД2 имеют немного больше, если вообще больше, жира в поджелудочной железе, чем те, у кого только непереносимость глюкозы.
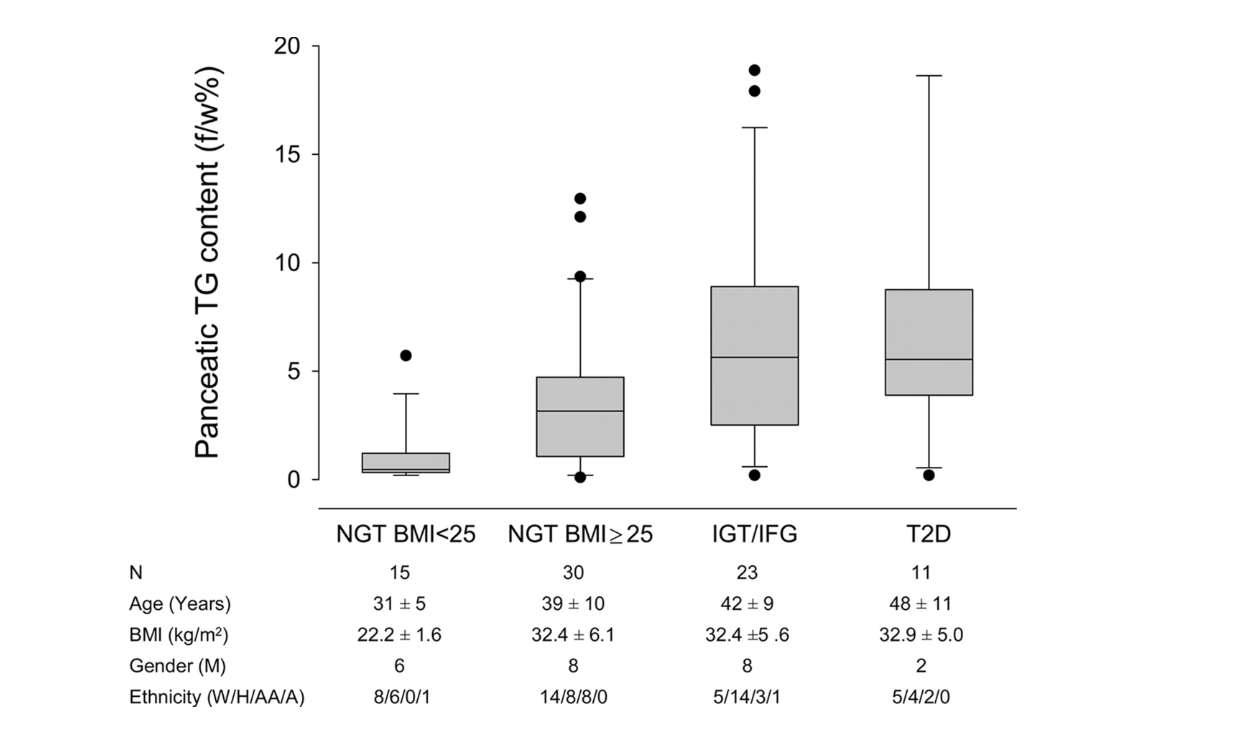
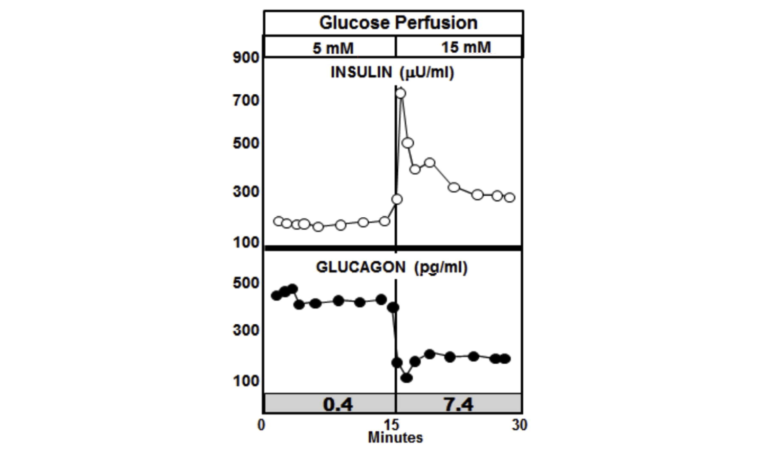
Рисунок 3: Паракринный ответ на увеличение перфузии глюкозы у нормальных животных без диабета
Теперь рассмотрим, что происходит, когда перфузия глюкозы утраивается в поджелудочной железе у диабетических крыс. На графике ниже наиболее заметной особенностью является отсутствие всплеска инсулина и отсутствие подавления глюкагона. Фактически, уровни инсулина увеличиваются у животных с диабетом до более высокого уровня, чем у животных без диабета, но глюкагон не подавляется и продолжает расти (10).
Соотношение инсулина к глюкагону, когда происходит всплеск инсулина, у недиабетических животных составляет около 7.4, тогда как такое соотношение у диабетических животных ниже 0.4.
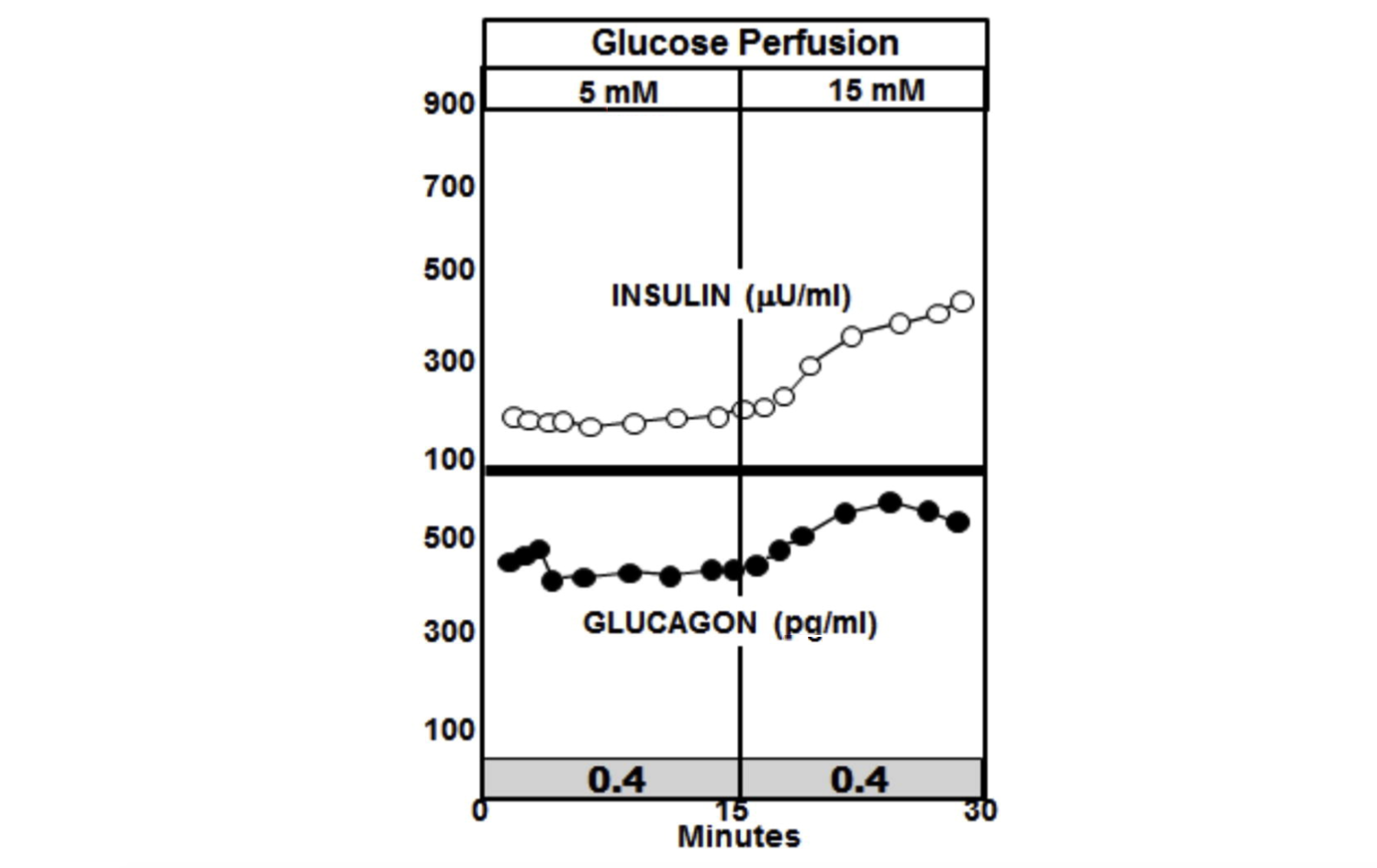
Рисунок 4: Перфузия глюкозы у крыс с диабетом
Отношение инсулина к глюкагону, равное 7.4, сигнализирует печени о том, что в крови содержится большое количество глюкозы, и печень должна начать ее извлекать и преобразовывать в гликоген. Отношение 0.4 посылает противоположный сигнал. Оно сообщает печени, что животное, несмотря на повышенный уровень глюкозы, истощается, и печени следует начать производить больше глюкозы и продолжать ее выделять. Все это приводит к хронически повышенным уровням глюкозы у пациентов с СД 2.
Но почему реакция инсулина притупляется у пациентов с СД2? Исследования показали, что как инсулин-продуцирующие бета-клетки, так и альфа-клетки, которые секретируют глюкагон, подвергаются риску, когда эктопический жир проникает в островки поджелудочной железы, где находятся эти клетки. Наряду с эктопическим жиром церамид, сфинголипид, еще больше препятствует функционированию этих клеток.
Эктопический жир попадает в различные органы, включая поджелудочную железу, при превышении PFT. Под влиянием серин-пальмитоилтрансферазы пальмитат (насыщенный жир, выделяемый печенью во время липогенеза de novo lipogenesis) соединяется с серином с образованием церамида, который в течение многих лет считается движущей силой инсулинорезистентности (11). Серин пальмитоилтрансфераза, ограничивающий скорость синтеза фермент в синтезе церамида, экспрессируется на более высоких уровнях у мышей db/db, склонных к диабету. По сравнению с +/+ мышами дикого типа, меченый церамид обнаруживается в гораздо больших количествах в плазме мышей db/db. Эксперименты, в которых культивируемые альфа-клетки подвергаются воздействию только одного инсулина или инсулина плюс церамид, генерируют заметно разные уровни продукции мРНК глюкагона. На приведенной ниже диаграмме видно, что без ингибирующего влияния инсулина (и без церамида) альфа-клетки продуцируют определенное количество глюкагона. При добавлении инсулина, как и ожидалось, количество мРНК глюкагона заметно падает. Но когда такое же количество инсулина добавляется вместе с церамидом, количество мРНК глюкагона, продуцируемого альфа-клетками, резко возрастает. Очевидно, что церамид в островках может вызвать резистентность к инсулину. Нарушение состояния бета-клеток из-за эктопической инфильтрации жира (липотоксичность), которая приводит к снижению выхода инсулина, в сочетании с устойчивостью к инсулину альфа-клеток, создает идеальный шторм для возникновения СД2 (12).
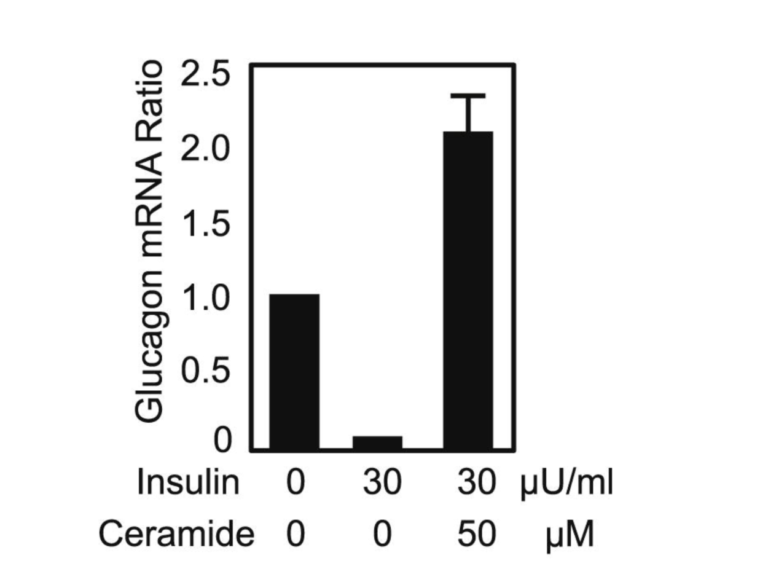 Рисунок 5: Соотношение мРНК глюкагона в культивируемых альфа-клетках при введении инсулина и церамида.
Рисунок 5: Соотношение мРНК глюкагона в культивируемых альфа-клетках при введении инсулина и церамида.
Единственный вывод, который может быть разумно сделан из результатов этих исследований, состоит в том, что удаление углеводов из рациона приводит к большей потере веса и большему снижению HbA1c. Логично, что на аверсе может показаться, что слишком много углеводов в рационе создает проблему.
Как и в случае с T1DM, настоящей проблемой является перепроизводство глюкагона. Мыши, у которых нокаутированны рецепторы глюкагона (GcgR -/-), не страдают ожирением или СД2.
Рада сообщить, что группа Вирта под руководством доктора Сары Холлберг использует высокожирную низкоуглеводную диету для обращения вспять СД2. http://galinaleb.com/dr-sarah-hallbergs-ted-talk/ , еще один пост http://galinaleb.com/avoid-carbs-not-fat/ . Г.Л.
 Д-ра. Майкл и Мэри Дэн Идес являются авторами 14 книг в области здравоохранения, питания и физических упражнений, включая бестселлер Protein Power.
Д-ра. Майкл и Мэри Дэн Идес являются авторами 14 книг в области здравоохранения, питания и физических упражнений, включая бестселлер Protein Power.
Доктор Майкл Идес родился в Спрингфилде, штат Миссури, и получил образование в Миссури, Мичигане и Калифорнии. Он получил степень бакалавра в области машиностроения в Калифорнийском государственном политехническом университете и степень доктора медицины в Университете Арканзаса. После завершения медицинского обучения и последипломного обучения он и его жена Мэри Дэн основали Medi-Stat Medical Clinics, сеть амбулаторных амбулаторных семейных клиник в центральном Арканзасе. С 1986 года доктор Майкл Идес работает в бариатрической, пищевой и метаболической медицине. Он и его жена занимались частной практикой, посвящая свое клиническое время исключительно бариатрической и диетологической медицине, получая непосредственный опыт лечения более 6000 человек, страдающих от высокого кровяного давления, диабета, повышенного уровня холестерина и триглицеридов и ожирения с их режимом питания.
Вместе супруги-врачи Eadеs дают многочисленные лекции широкой общественности и различным общественным организациям о своих методах лечения. Они оба были приглашенными экспертами по питанию на более чем 150 радио и телевизионных шоу, включая национальные сегменты для FOX и CBS.
References
- Sharpey-Schäfer EA. The Endocrine Organs: An introduction to the study of internal secretion. London: Longmans, Green, 1916.
- Lee Y, Berglund ED, Wang MY, Fu X, Yu X, Charron MJ, Burgess SC, Unger RH. Metabolic manifestations of insulin deficiency do not occur without glucagon action. Proc Natl Acad Sci USA. 109.37(2012): 14972-6.
- Boutens L, Stienstra R. Adipose tissue macrophages: going off track during obesity. Diabetologia. 59.5(2016): 879-94.
- Taylor R, Holman RR. Normal weight individuals who develop type 2 diabetes: the personal fat threshold. Clin Sci (Lond). 128.7(2015): 405-10.
- Unger RH, Scherer PE. Gluttony, sloth and the metabolic syndrome: a roadmap to lipotoxicity. Trends Endocrinol Metab. 21.6(2010): 345-52.
- Unger RH. Lipotoxic diseases. Annu Rev Med. 53(2002): 319-36.
- Unger RH. Minireview: weapons of lean body mass destruction: the role of ectopic lipids in the metabolic syndrome. Endocrinology. 144.12(2003): 5159-65.
- Shimabukuro M, Zhou YT, Levi M, Unger RH. Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA. 95.5(1998): 2498-502.
- Lingvay I, Esser V, Legendre JL, Price AL, Wertz KM, Adams-Huet B, Zhang S, Unger RH, Szczepaniak LS. Noninvasive quantification of pancreatic fat in humans. J Clin Endocrinol Metab. 94.10(2009): 4070-6.
- Unger RH, Roth MG. A new biology of diabetes revealed by leptin. Cell Metab. 21.1(2015): 15-20.
- Chavez JA, Summers SA. A ceramide-centric view of insulin resistance. Cell Metab. 15.5(2012): 585-94.
- Lee Y, Berglund ED, Yu X, Wang MY, Evans MR, Scherer PE, Holland WL, Charron MJ, Roth MG, Unger RH. Hyperglycemia in rodent models of type 2 diabetes requires insulin-resistant alpha cells. Proc Natl Acad Sci USA. 111.36(2014): 13217-22.
- Schwarz JM, Neese RA, Turner S, Dare D, Hellerstein MK. Short-term alterations in carbohydrate energy intake in humans. Striking effects on hepatic glucose production, de novo lipogenesis, lipolysis, and whole-body fuel selection. J Clin Invest. 96.6(1995): 2735-43.
- Brown MS, Goldstein JL. Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 7.2(2008): 95-6.
- Mendez CE, Walker RJ, Eiler CR, Mishriky BM, Egede LE. Insulin therapy in patients with type 2 diabetes and high insulin resistance is associated with increased risk of complications and mortality. Postgrad Med. 1-7(2019).
- Feinman RD, Pogozelski WK, Astrup A, Bernstein RK, Fine EJ, Westman EC, Accurso A, Frassetto L, Gower BA, McFarlane SI, et al. Dietary carbohydrate restriction as the first approach in diabetes management: critical review and evidence base. Nutrition. 31.1(2015): 1-13.
